【数据挖掘】使用R语言进行聚类分析
本文主要介绍在R语言中使用k-means和K-Medoids进行聚类分析的方法。
一、首先介绍下聚类分析中主要的算法:
K-均值聚类(K-Means) 十大经典算法
K-中心点聚类(K-Medoids)
密度聚类(DBSCAN)
系谱聚类(HC)
期望最大化聚类(EM) 十大经典算法
| 聚类算法 | 软件包 | 主要函数 |
|---|---|---|
| K-means | stats | kmeans() |
| K-Medoids | cluster | pam() |
| 系谱聚类(HC) | stats | hclust(), cutree(), rect.hclust() |
| 密度聚类(DBSCAN) | fpc | dbscan() |
| 期望最大化聚类(EM) | mclust | Mclust(), clustBIC(), mclust2Dplot(), densityMclust() |
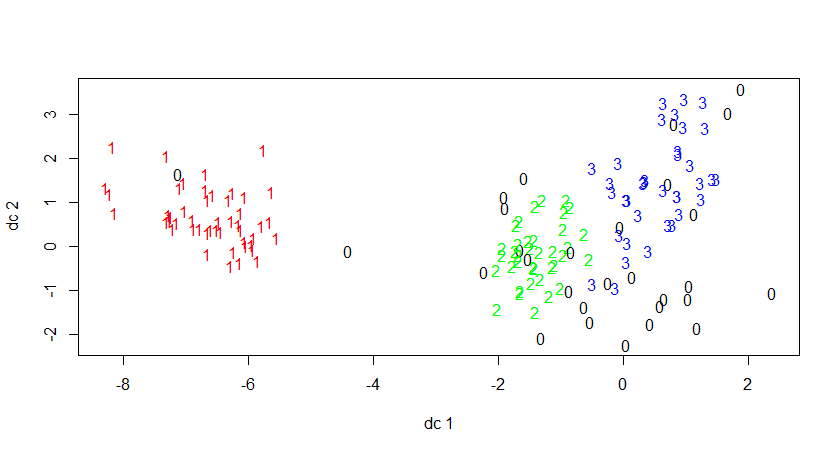
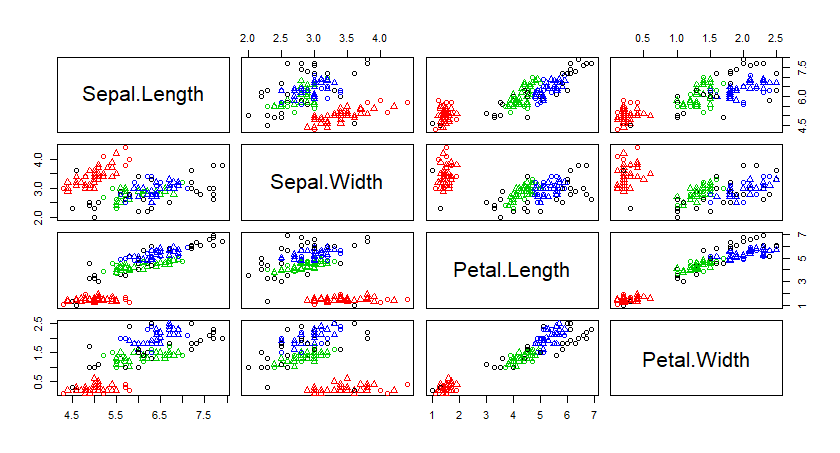
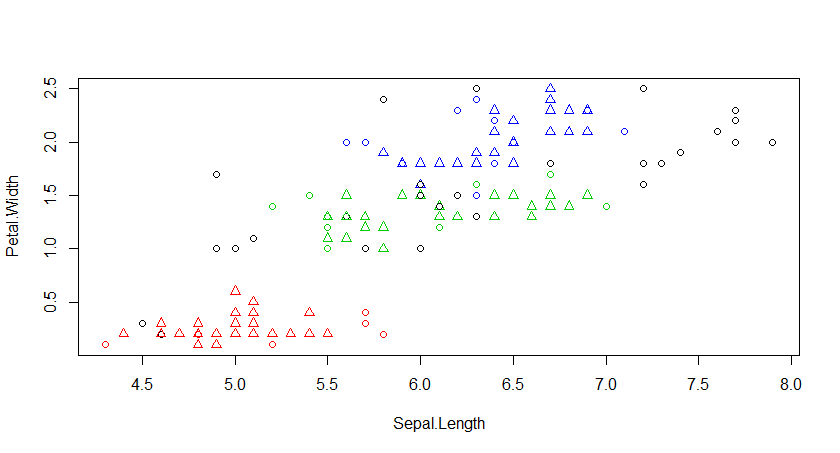
二、用iris数据集进行kmeans分析
1 | |
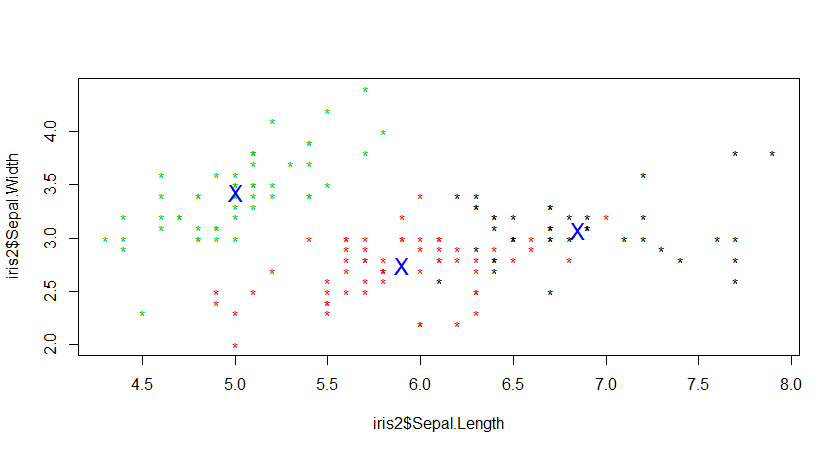
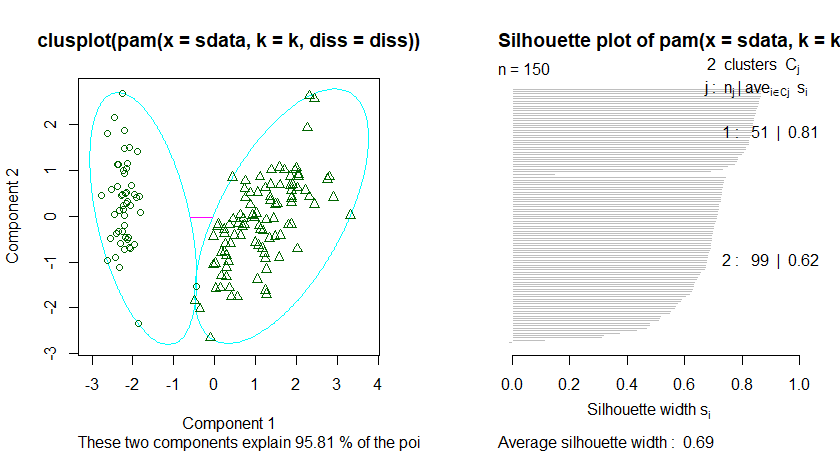
三、使用K-Mediods 进行聚类分析
K-Mediods函数跟Kmeans函数基本类似,不同的是,Kmeans是选择簇中心来表示聚类簇,而K-Mediods选择靠近簇中心的对象来表示聚类簇。在含有离群点的情况,下K-Mediods的鲁棒性(稳定性)要更好。
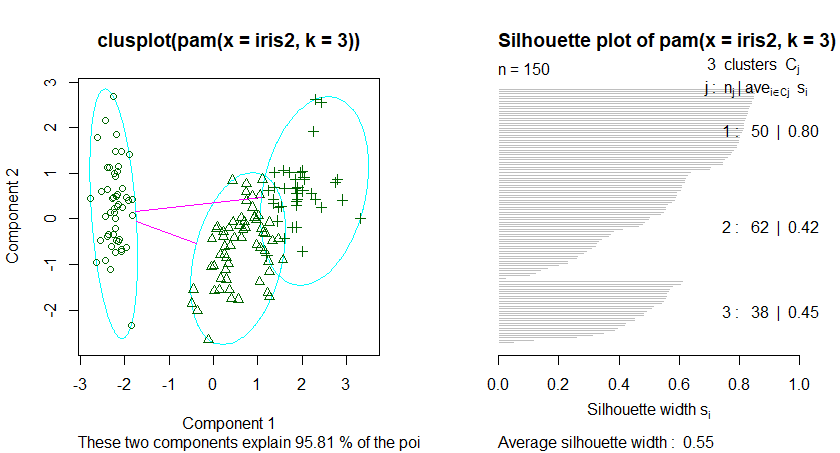
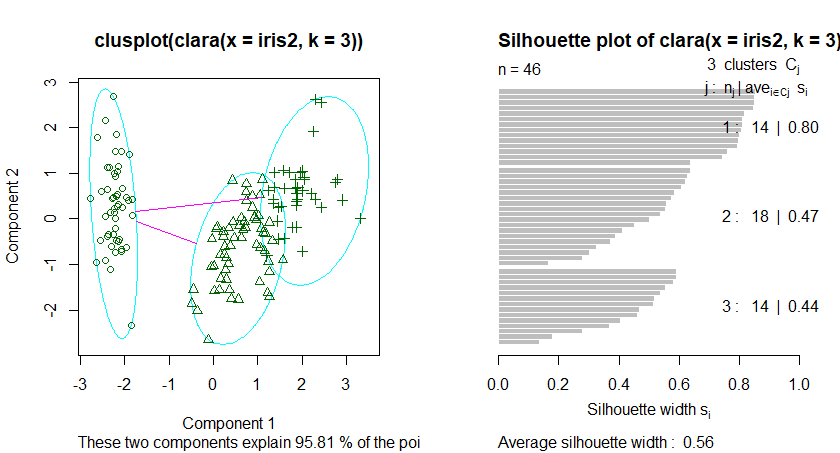
基于中心点的划分算法PAM是K-Mediods中的经典算法,但是PAM很难扩展到较大数据集上,而Clara算法是对PAM算法的改进,他是在较大数据集中分为几个小数据集,分别进行PAM算法,并返回最好的聚类。因此在处理较大数据集的情况下CLARA算法要优于PAM算法,
在R的cluster包中的PAM和CLARA函数分别实现了上述两个算法,但是这两个函数都需要用户指定k值,即中心点的个数。fpc包中的pamk()函数提供了更加强大的算法,该函数不要求用户输入k值,而是自动调用pam或者clara来根据最优平均阴影宽度来估计聚类簇个数来划分数据集。
K-mediods算法描述
a) 首先随机选取一组聚类样本作为中心点集
b) 每个中心点对应一个簇
c) 计算各样本点到各个中心点的距离(如欧几里德距离),将样本点放入距离中心点最短的那个簇中
d) 计算各簇中,距簇内各样本点距离的绝度误差最小的点,作为新的中心点
e) 如果新的中心点集与原中心点集相同,算法终止;如果新的中心点集与原中心点集不完全相同,返回b)
1 | |
1 | |
1 | |
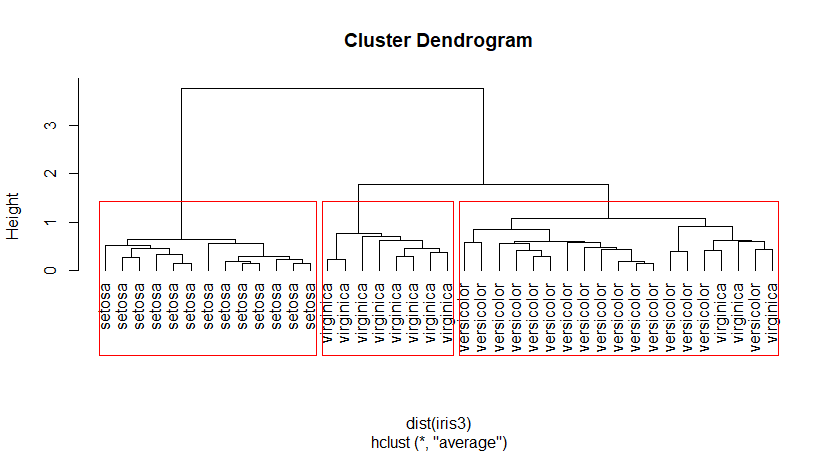
四、层次聚类HCluster
基本思想:
1、开始时,将每个样本作为一类。
2、规定某种度量作为样本之间距离以及类距离之间的度量,并且计算之。(hculster里边的dist方法以及method属性)
3、将距离最短的两个类合并为一个类。
4、重复2-3,即不断合并最近的两个类,每次减少一个类,直到所有的样本合并为一个类。
点与点的距离和类与类之间距离的计算可以参考R-modeling关于距离的介绍。
1 | |
五、基于密度的聚类
前边的k-Means和k-Mediods算法比较适用于簇为球型的,对于非球型的,一般需要基于密度的聚类,比如DBSCAN
要了解DBSCAN,我们需要知道几个基本概念
r-邻域:给定点半径为r的区域。
核心点:如果一个点的r邻域内最少包含M个点,则该点称为核心点。
直接密度可达:对于核心点P而言,如果另一个点O在P的r邻域内,那么称O为P的直接密度可达点。
密度可达:对于P的直接密度可达点O的r邻域内,如果包含另一个点Q,那么称Q为P的密度可达点。
密度相连:如果Q和N都是核心点P的密度可达点,但是并不在一条直线路径上,那么称两者为密度相连。
算法思想:
- 指定R和M。
- 计算所有的样本点,如果点p的r邻域内有超过M个点,那么创建一个以P为核心点的新簇。
- 反复寻找这些核心点的直接密度可达点(之后可能是密度可达),将其加入到相应的簇,对于核心点发生密度相连的情况加以合并。
- 当没有新的点加入到任何簇中时,算法结束。
优点:
(1)聚类速度快且能够有效处理噪声点和发现任意形状的空间聚类;
(2)与K-MEANS比较起来,不需要输入要划分的聚类个数;
(3)聚类簇的形状没有偏倚;
(4)可以在需要时输入过滤噪声的参数。
缺点:
(1)当数据量增大时,要求较大的内存支持I/O消耗也很大;
(2)当空间聚类的密度不均匀、聚类间距差相差很大时,聚类质量较差,因为这种情况下参数MinPts和Eps选取困难
(3)算法聚类效果依赖与距离公式选取,实际应用中常用欧式距离,对于高维数据,存在“维数灾难”。
R语言中的例子:
1 | |
1 | |
1 | |